Blog
17 April 2024
Shining a light on Haemophilia: An Interview with Assoc. prof. Maria Hulikova, Unilabs Genetics
Haemophilia is a rare genetic bleeding disorder that affects the blood's ability to clot properly, leading to prolonged bleeding and/or spontaneous internal bleeding. World Haemophilia Day provides an opportunity to raise awareness about this condition and the challenges faced by individuals living with it. At Unilabs, we are committed to advancing diagnostics and understanding of haemophilia. Today, we have the privilege of speaking with Assoc. prof. Maria Hulikova, a distinguished haematologist from Centre for Haemostasis and Thrombosis at Unilabs, to explore the complexities of haemophilia and the role of diagnostics in its management.
What are some common symptoms or signs that might indicate a potential case of haemophilia, and how important is early diagnosis in managing the condition?
The main clinical symptom of haemophilia is bleeding. The extent and intensity of bleeding depend on the coagulation activity of deficient Factor VIII. Individuals with mild (FVIII more than 5%) and moderate (FVIII more than 1% and less than 5%) forms of haemophilia do not experience many problems. They do not have spontaneous bleeding and are essentially unaware of being ill. They only face an increased risk of bleeding during injuries, accidents, tooth extractions, surgeries, or invasive procedures (biopsies). Typical spontaneous bleeding without an apparent trigger is seen in patients with severe haemophilia (FVIII less than 1%). Patients with severe haemophilia often suffer from frequent, recurrent bleeding into joints (80-90%), muscles, and soft tissues (10-20%, making intramuscular injections contraindicated), as well as skin, mucous membranes, haematuria (blood in urine), melena (blood in stool), and potentially life-threatening bleeding into internal organs (brain, lungs, digestive tract). Approximately 90% of bleeding episodes in haemophilia patients involve joints and muscles, with the knee and elbow joints being the most affected. Joint bleeding manifests with symptoms of pain, swelling, and restricted mobility. Recurrent joint bleeding leads to structural damage, cartilage loss, subchondral cysts, osteophyte formation, joint fibrosis, ankylosis, irreversible joint dysfunction, immobility, and eventual disability. Concurrently, muscle atrophy worsens, accompanied by contractures and joint axis deviations. Early diagnosis of haemophilia enables prompt and adequate substitution of coagulation factors, prophylactic administration to prevent recurrent bleeding, and permanent joint damage.
Given the genetic nature of haemophilia, what role do genetic tests play in diagnosing and predicting the severity of the condition?
For the diagnosis of haemophilia, a bleeding history (personal and familial, especially from the maternal side) is essential. In laboratory findings, haemophilia is characterised by prolonged activated partial thromboplastin time (aPTT), which extends with a decrease in FVIII below approximately 30%. Confirmation of the diagnosis involves determining the coagulation activity of FVIII. Depending on the severity of the FVIII deficiency, haemophilia is classified into severe (less than 1%), moderate (1 - 5%), and mild (more than 5 - 40%) forms. Molecular biological testing is part of haemophilia diagnosis, identifying the causal mutation through DNA analysis of FVIII genes. DNA diagnosis clarifies the extent and localisation of the genetic disorder, reliably detects carriers, and is essential for prenatal foetal diagnostics. Prenatal examination during pregnancy involves chorionic villus biopsy at 11-14 weeks of gestation or alternatively, amniocentesis at 15-18 weeks of gestation.
How have advancements in diagnostic technologies improved our ability to diagnose and monitor haemophilia over the years?
In recent years, significant progress has been made in haemophilia diagnosis through the introduction of genetic tests, DNA analysis of FVIII genes, and prenatal examination of at-risk mothers. In laboratory coagulation tests, prolonged activated partial thromboplastin time (aPTT) is observed, while other tests such as prothrombin time, thrombin time, fibrinogen, and platelets remain normal. Coagulation and chromogenic methods are currently used to diagnose and classify haemophilia patients based on FVIII activity. The one-stage coagulation method is based on aPTT using plasma deficient in FVIII. The two-stage coagulation method is based on determining the activity of FV and FX, which are generated in amounts directly proportional to the activity of FVIII in the sample under investigation. The chromogenic method has opened a new path for determining FVIII concentrate, with a principle similar to the two-stage test. The amount generated by FX is directly proportional to the activity of FVIII. The two-stage coagulation method and chromogenic method are the reference methods of the European Pharmacopoeia for determining FVIII in coagulation factor concentrates for haemophilia treatment.
Haemophilia management often requires a multidisciplinary approach. How does Unilabs collaborate with other healthcare professionals to provide comprehensive care for individuals with haemophilia?
The management of haemophilia has seen significant advancements throughout its history. Treating haemophilia was practically impossible in the past. However, today, what was once considered an incurable disease is now treatable.
A significant breakthrough occurred in the 1960s when plasma-derived coagulation factor VIII concentrates (CCF) began to be manufactured. Since 1992, recombinant coagulation factors (rCF) have been developed. These are produced through genetic engineering and serve as a safe and effective alternative to activated plasma concentrates. The production process of rCF has undergone gradual refinement and enhancement, with a current preference for fourth generation factors with extended action.
Currently, there is also what's known as non-factor therapy available, which uses an alternative hemostatic agent called Emicizumab for replacing the coagulation factor. The wide range of products reflects the evolution of haemophilia treatment. The current therapeutic options in its treatment are listed in the table below:
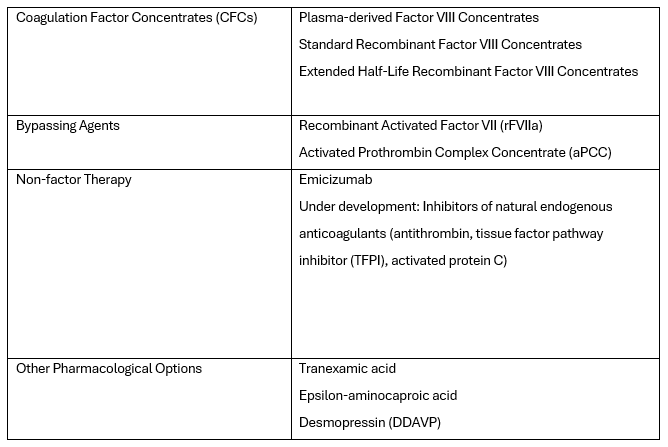
In the past, the treatment of haemophilia relied on substituting the missing factor VIII to stop bleeding. Currently, the World Federation of Haemophilia (WFH) prefers and recommends regular long-term prophylaxis, which is effective in preventing haemarthrosis and other spontaneous and breakthrough bleeds, maintaining musculoskeletal health, and improving the quality of life. WFH recommends initiating prophylaxis with standard or extended half-life clotting factor concentrates or other haemostatic agents before the onset of joint disease and ideally before reaching three years of age. The goal of prophylaxis is to prevent any bleeding episodes in patients, achieving what is known as zero bleeding.
Management and prevention of haemophilia require a multidisciplinary approach, involving collaboration with geneticists for accurate diagnosis of the congenital bleeding disorder - haemophilia, orthopaedists for the treatment and prevention of haemarthrosis, haemophilic arthropathy, and physiotherapists for maintaining musculoskeletal health and improving the quality of life of patients with haemophilia.